In this notebook we will explore how to run an RNA-seq analysis using DESeq2. At the end of this section you should have a reasonable grasp of:
How to create a DESeq2 object
How to identify potential confounding variables
How to run DESeq2 with a specific design formula
How to read and interpret DESeq2 results
How to run a GSEA analysis
As differential gene expression (DGE) involves cross-sample comparisons, it is essential to use normalized counts. RPKM, FPKM and TMM remove most noise affiliated with sequencing depth as discussed previously. However, these approaches only deal with absolute counts and perform poorly when the transcript distributions are skewed due to highly and differentially-expressed genes. This concern is managed by methods such as DESeq and others which ignore highly variable and/or expressed genes. We also need to account for differences in transcript length across samples/conditions, positional biases along transcript (Cufflinks), and gene-specific GC content (EDAseq). NOISeq can be used to create visualizations for all these biases and efficiently decide what normalization tool should be used. Despite these steps, additional batch effects can still occur and should be minimized with good experimental design or with batch correction methods like COMBAT or ARSyN.
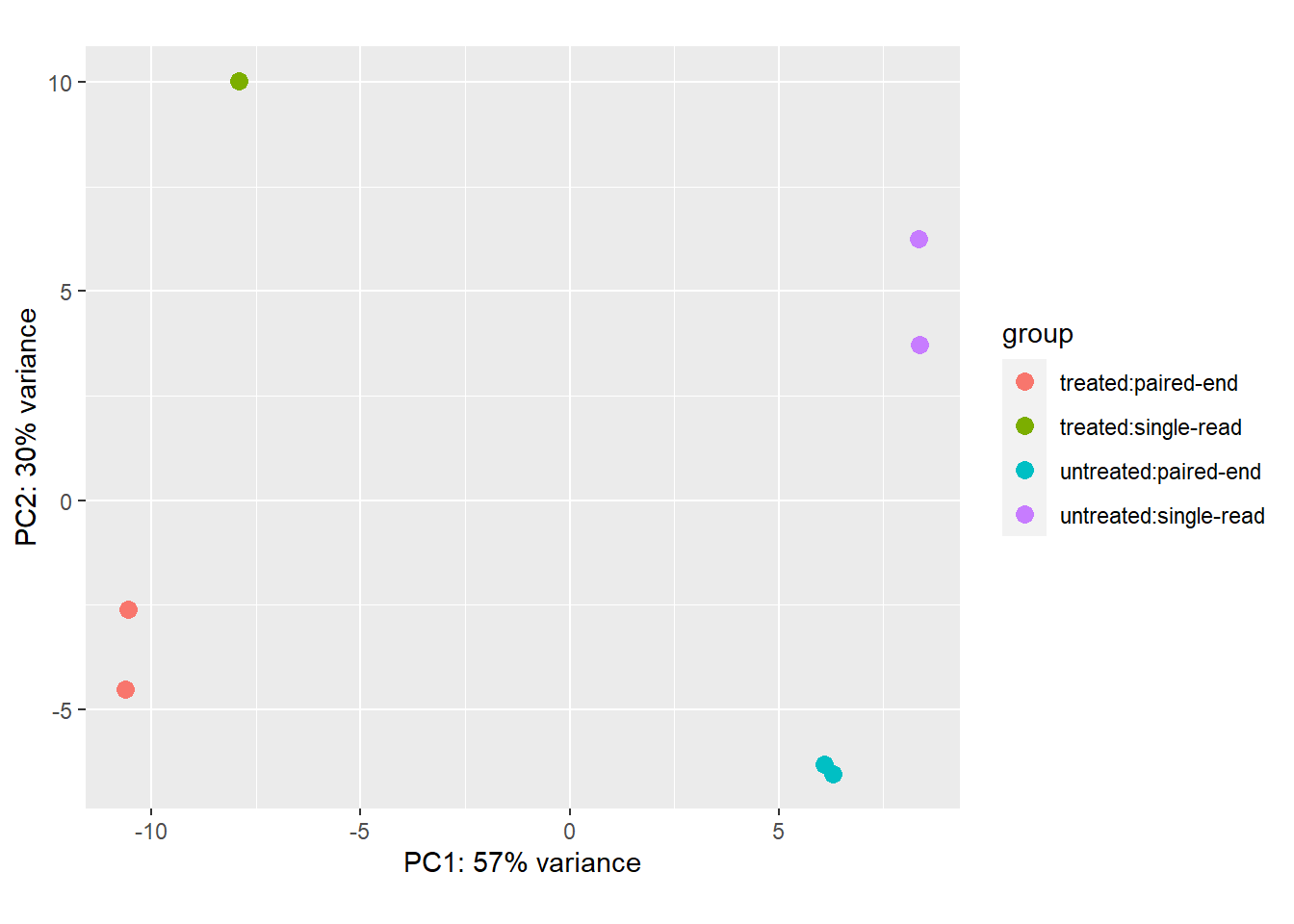
As RNA-seq data is expressed as counts, most differential expression methods model the counts with discrete distributions such as the poisson, or the negative-binomial (gamma-poisson) which allows for additional variance (overdispersion) beyond what is expected from sampling a random pool of molecules. In practice, a discrete distribution is not required if we account for the sampling variance of small read counts (particularly in studies with few replicates). Many methods that transform normalized RNA-seq counts and learn the underling variance structure perform quite well. It is also worth noting that with sufficient normalization, the data will no longer hold its discrete property and should be modeled with a continuous distribution. In practice, most researchers use one of edgeR, DESeq2 or limma. All three methods have variants that allow for the inclusion of covariates (worth using based on PCA plots).
5.2 1. Loading Count Matrix into R
The first step to a DESeq2 analysis is to load your data into R and create a DESeq2 object. We need two items:
a count matrix with genes as rows and samples as columns
a metadata matrix (colData) which contains the condition and covariate data for all samples
set.seed(1234)library(DESeq2)
Loading required package: S4Vectors
Loading required package: stats4
Loading required package: BiocGenerics
Attaching package: 'BiocGenerics'
The following objects are masked from 'package:stats':
IQR, mad, sd, var, xtabs
Welcome to Bioconductor
Vignettes contain introductory material; view with
'browseVignettes()'. To cite Bioconductor, see
'citation("Biobase")', and for packages 'citation("pkgname")'.
Attaching package: 'Biobase'
The following object is masked from 'package:MatrixGenerics':
rowMedians
The following objects are masked from 'package:matrixStats':
anyMissing, rowMedians
library(tidyverse)
Warning: package 'ggplot2' was built under R version 4.2.3
Warning: package 'tibble' was built under R version 4.2.3
Warning: package 'dplyr' was built under R version 4.2.3
using 'apeglm' for LFC shrinkage. If used in published research, please cite:
Zhu, A., Ibrahim, J.G., Love, M.I. (2018) Heavy-tailed prior distributions for
sequence count data: removing the noise and preserving large differences.
Bioinformatics. https://doi.org/10.1093/bioinformatics/bty895
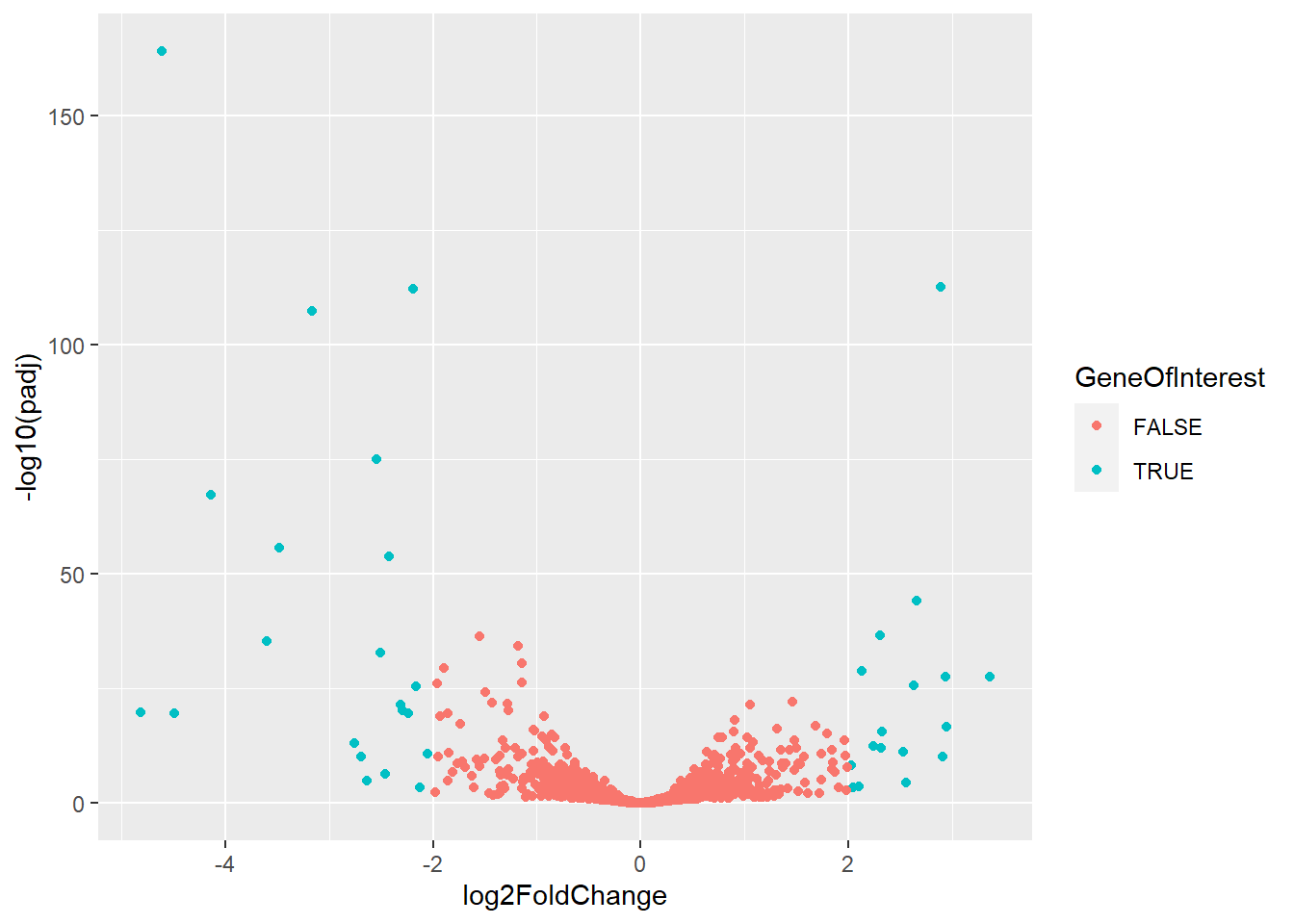
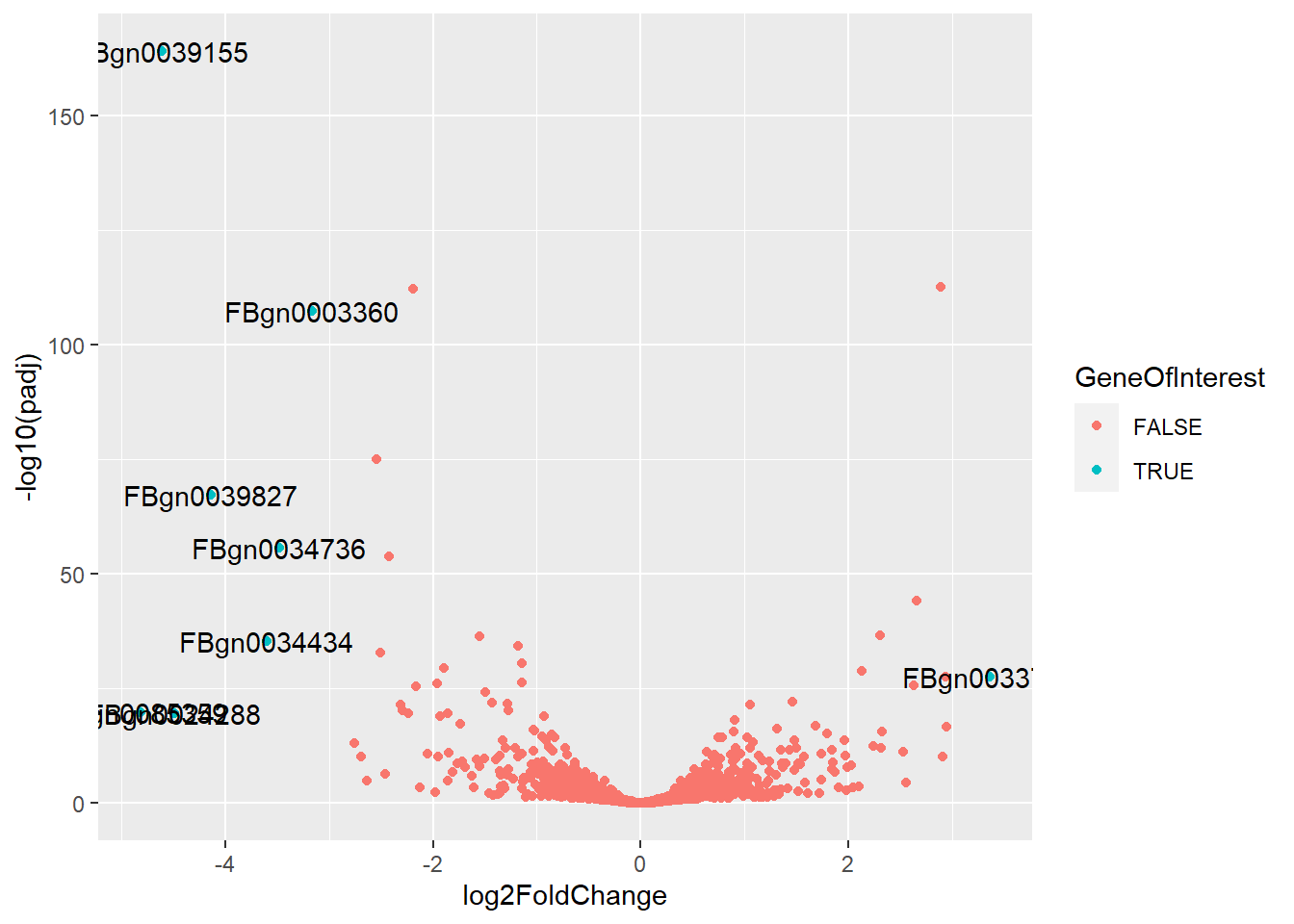
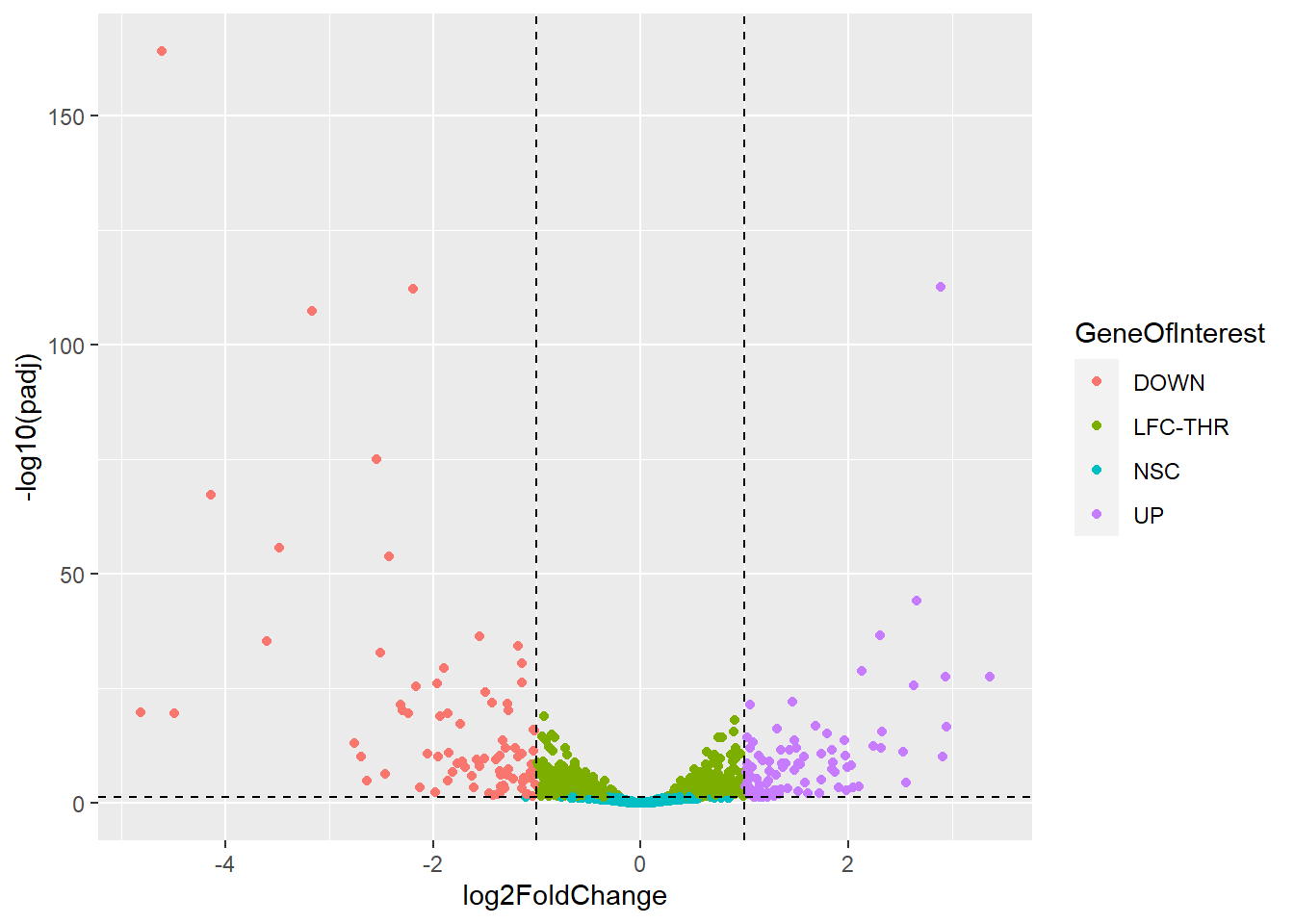
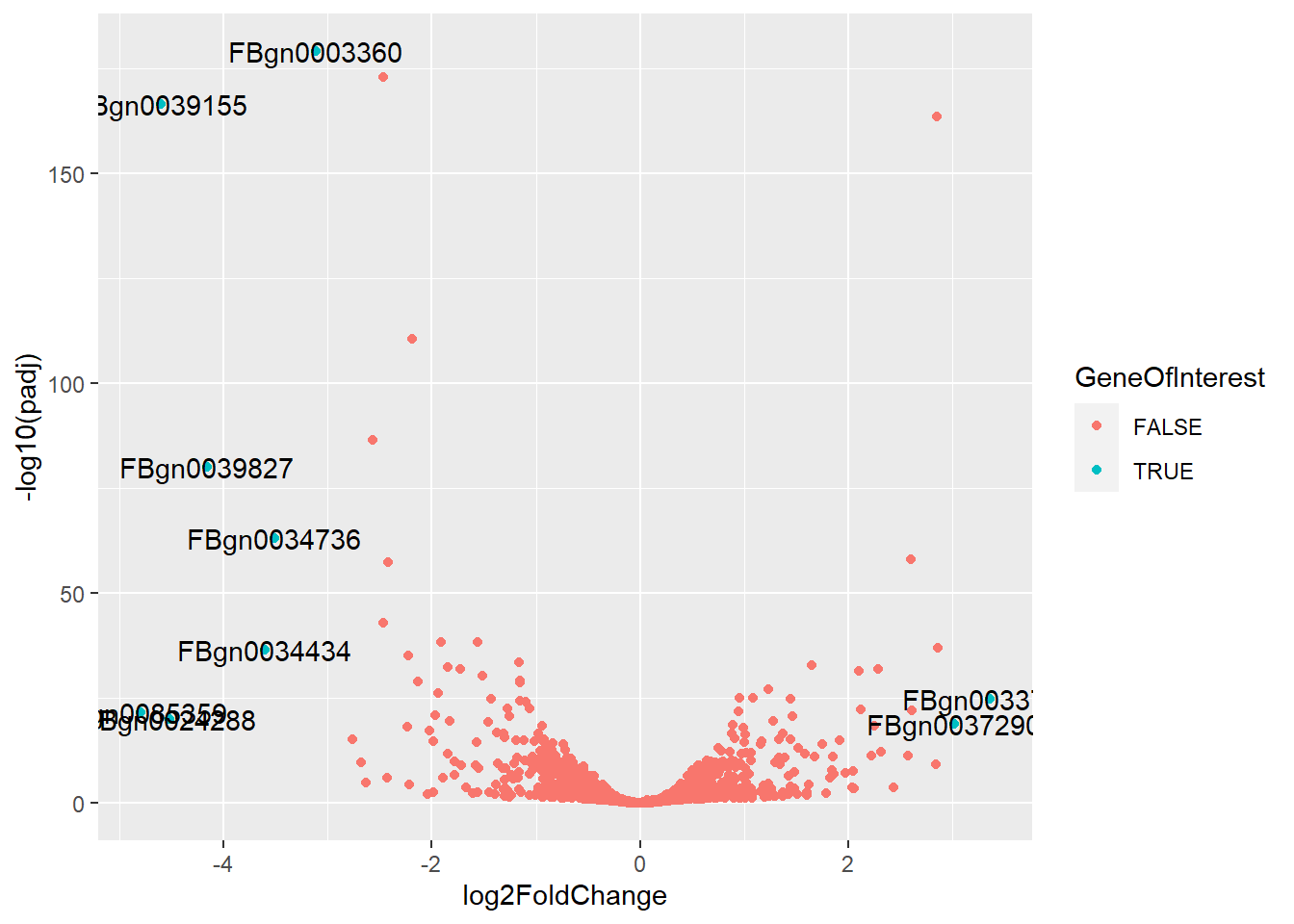
#Add Name of Genes if they are a gene of interestex_res <- df_resex_res$GeneID <-rownames(ex_res)ex_res$GeneOfInterest <- ex_res$padj <0.05&abs(ex_res$log2FoldChange) >=3ggplot(ex_res, aes(x = log2FoldChange, y =-log10(padj))) +geom_point(aes(color = GeneOfInterest)) +geom_text(data = ex_res %>%filter(GeneOfInterest), aes(label = GeneID))
Note: levels of factors in the design contain characters other than
letters, numbers, '_' and '.'. It is recommended (but not required) to use
only letters, numbers, and delimiters '_' or '.', as these are safe characters
for column names in R. [This is a message, not a warning or an error]
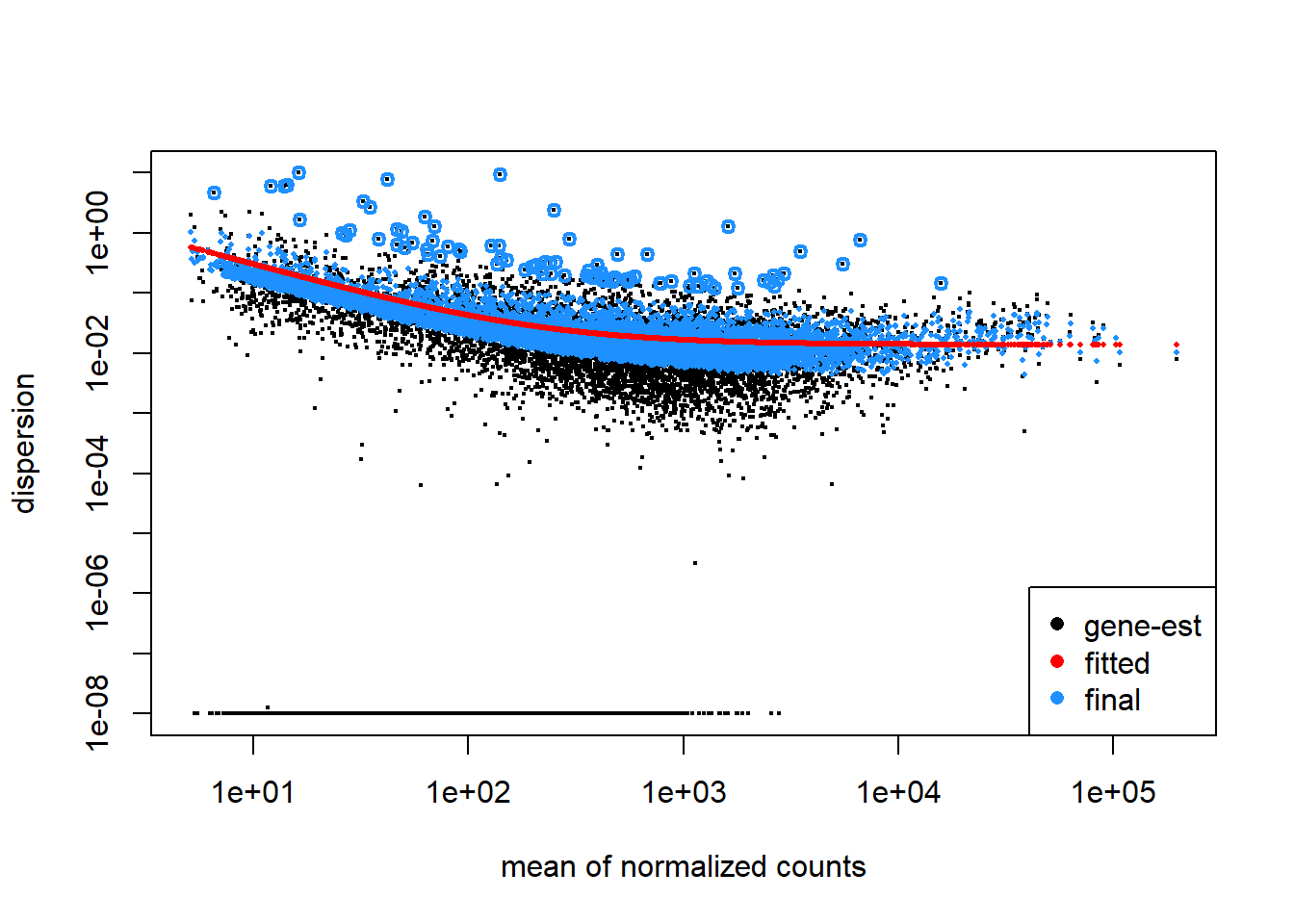
codds <-DESeq(codds)
using pre-existing size factors
estimating dispersions
found already estimated dispersions, replacing these
gene-wise dispersion estimates
mean-dispersion relationship
Note: levels of factors in the design contain characters other than
letters, numbers, '_' and '.'. It is recommended (but not required) to use
only letters, numbers, and delimiters '_' or '.', as these are safe characters
for column names in R. [This is a message, not a warning or an error]
final dispersion estimates
Note: levels of factors in the design contain characters other than
letters, numbers, '_' and '.'. It is recommended (but not required) to use
only letters, numbers, and delimiters '_' or '.', as these are safe characters
for column names in R. [This is a message, not a warning or an error]
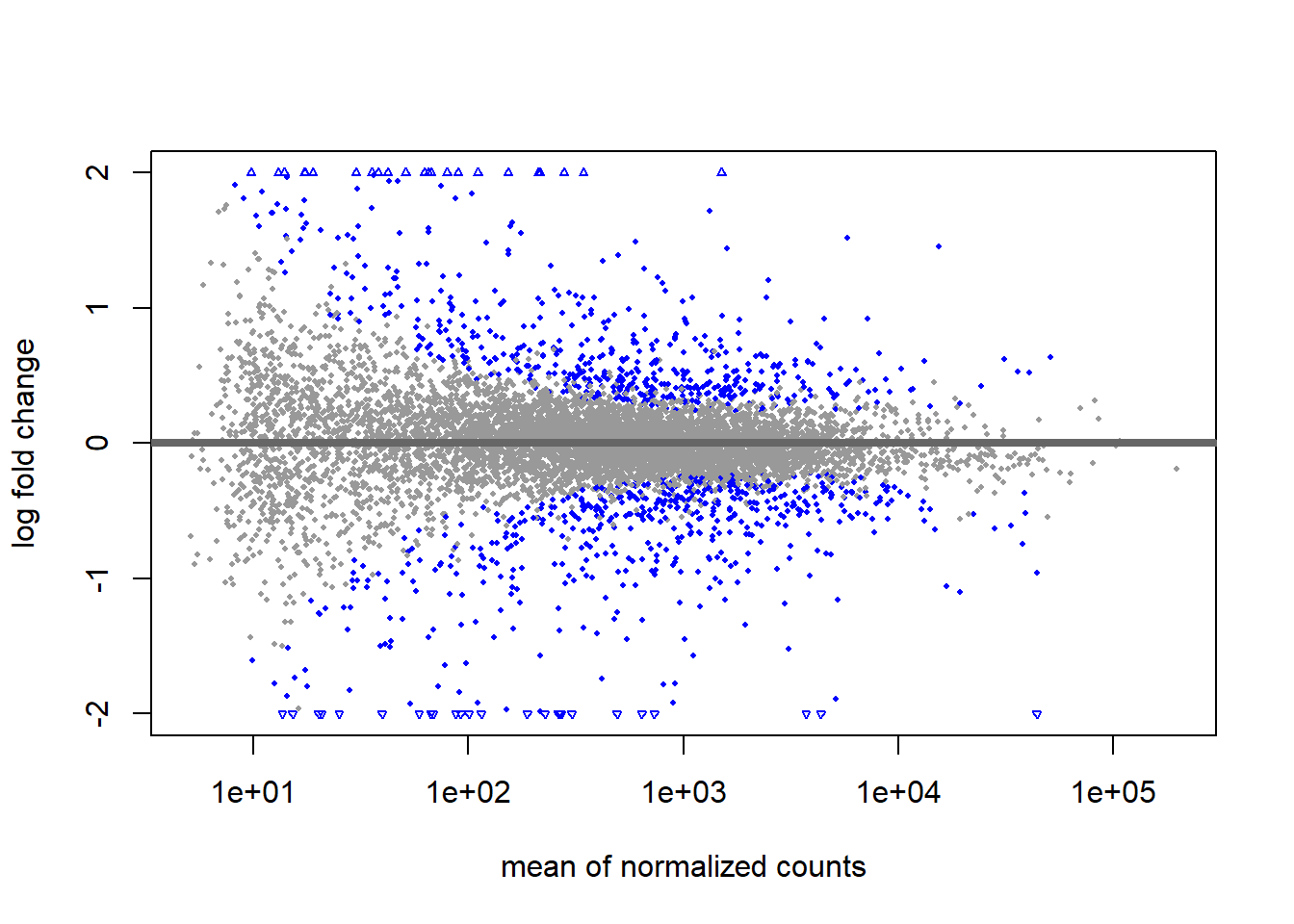
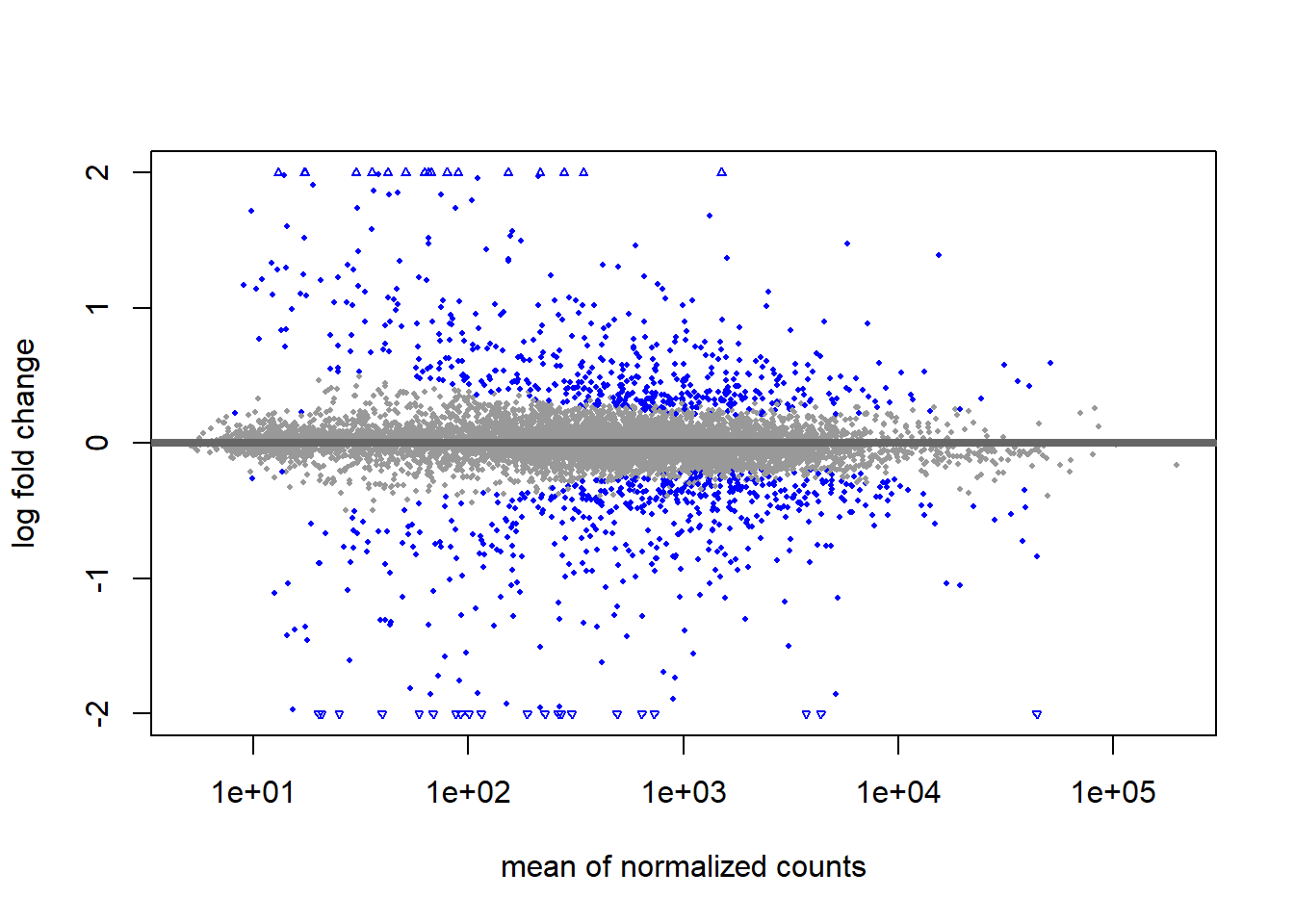
using 'apeglm' for LFC shrinkage. If used in published research, please cite:
Zhu, A., Ibrahim, J.G., Love, M.I. (2018) Heavy-tailed prior distributions for
sequence count data: removing the noise and preserving large differences.
Bioinformatics. https://doi.org/10.1093/bioinformatics/bty895
Data and code from Stephen Turner (stephenturner.github.io).
Dataset includes 8 smooth muscle cell samples from 4 different cell lines and 2 distinct treatments. It represents the outputs from DESeq2 using the following formula: ~ cell line + treatment.
library(fgsea)#Load DESeq2 resultsres <-read.csv("../Data/deseq-results-tidy-human-airway.csv",row.names ='X')#Convert to Gene Symbollibrary(org.Hs.eg.db)
Loading required package: AnnotationDbi
Attaching package: 'AnnotationDbi'
The following object is masked from 'package:dplyr':
select
#Get Gene Set (MSigDB Hallmarks)pathways.hallmark <-gmtPathways("../Data/h.all.v2023.2.Hs.symbols.gmt")#Run GSEAfgseaRes <-fgsea(pathways=pathways.hallmark, stats=ranks, nperm=1000)
Warning in fgsea(pathways = pathways.hallmark, stats = ranks, nperm = 1000):
You are trying to run fgseaSimple. It is recommended to use fgseaMultilevel. To
run fgseaMultilevel, you need to remove the nperm argument in the fgsea
function call.
Warning in preparePathwaysAndStats(pathways, stats, minSize, maxSize, gseaParam, : There are ties in the preranked stats (17.23% of the list).
The order of those tied genes will be arbitrary, which may produce unexpected results.
Exercise: Repeat the above using a gene set you found on the MSigDB webpage.
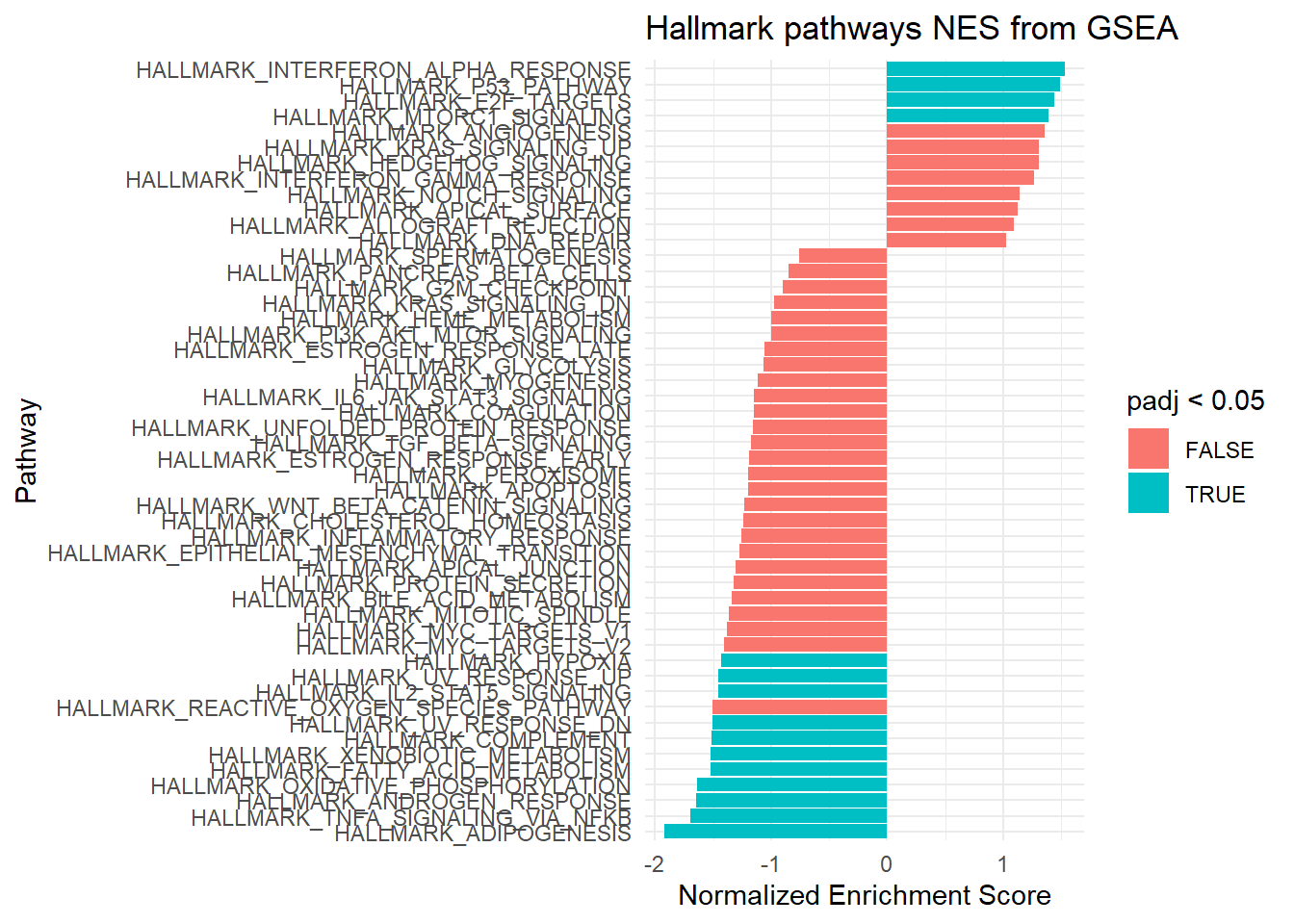
fgseaResTidy <- fgseaRes %>%as_tibble() %>%arrange(desc(NES))# Show in a nice table:fgseaResTidy %>% dplyr::select(-leadingEdge, -ES, -nMoreExtreme) %>%arrange(padj)
ggplot(fgseaResTidy, aes(reorder(pathway, NES), NES)) +geom_col(aes(fill=padj<0.05)) +coord_flip() +labs(x="Pathway", y="Normalized Enrichment Score",title="Hallmark pathways NES from GSEA") +theme_minimal()
5.7 5. Incorporating other Genomic assays
5.7.1 ATAC-seq
Chromatin Accessibility can be used in tandem with gene expression to identify cis-regulatory elements (CREs) that affect transcription.
5.7.2 Whole Genome Sequencing (WGS)
Identifying SNPs for each RNA-seq sample can be used to identify expression quantitative trait loci (eQTLs). These represent individual variants that have a significant effect on the expression level of a gene. Consider looking at the GTEx Portal for a large suite of eQTL studies.
5.7.3 DNA Methylation (WGBS)
Differentially methylated sites could help explain transcription patterns found in RNA-seq.